
Computational Chemistry for Molecular Building Blocks
Understanding structural and electronic properties of nano-scaled systems is an important prerequisite for further development of these systems. Besides experimental methods, quantum chemical computations are crucial to achieve a better understanding. On the one hand, properties such as various types of optical spectra and energies can be calculated for an experimentally known molecular structure. On the other hand, structures of molecules or clusters may be determined by the interplay of calculated and experimental data. Moreover, calculations of systematic series of nano-particles with different size and composition provide data for model building. Computer programs are continuously developed further towards higher efficiency and accuracy, and towards new applications.
- Building principles of metal clusters with genetic algorithms in connection with density functional theory
- Investigation of binary clusters with perturbation-theory approaches
- Support of other INT research units concerning structure clarification and bonding characteristics
- Quantum chemical investigation of adsorption of organic molecules on metal surfaces with cluster and slab calculations
- Calculation of magnetic coupling parameters of bridged transition metal clusters based on ab-initio data and simulation of magnetic susceptibility
- Quantum chemical studies of systems in electronically excited states
- Further development of the program system TURBOMOLE (spin-orbit interactions, RI methods, basis sets) and other programs for the provision of special tools for the above-mentioned problem
- Elucidation of reaction mechanisms and computation of rate constants
Optical spectroscopy
Willem Klopper
We compute the electronic structure of ground- as well as electronically excited states of molecular building blocks of nano-scaled systems.
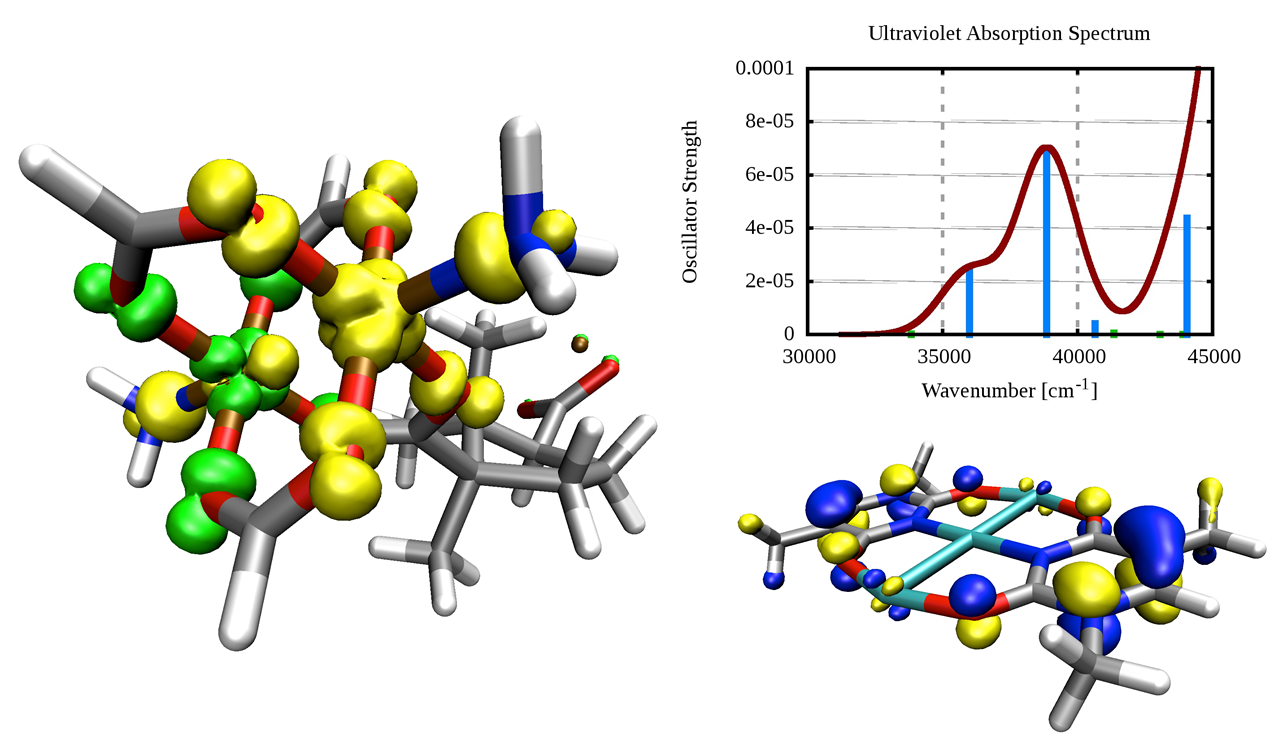
Spin density of an antiferromagnetically coupled copper-based paddle-wheel, which is used as secondary building unit in a metal-organic framework (left), and highest occupied molecular orbital of a 1-methylthymine-stabilized cationic trinuclear silver cluster together with its computed ultraviolet absorption spectrum (right).
K. D. Vogiatzis, W. Klopper, A. Mavrandonakis, and K. Fink
Magnetic properties of paddlewheels and trinuclear clusters with exposed metal sites
ChemPhysChem 12, 3307-3319 (2011)
Solid state electronic structure theory
Karin Fink
We perform embedded cluster calculations and periodic super cell calculations on the adsorption of molecules on metal and metal oxide surfaces.
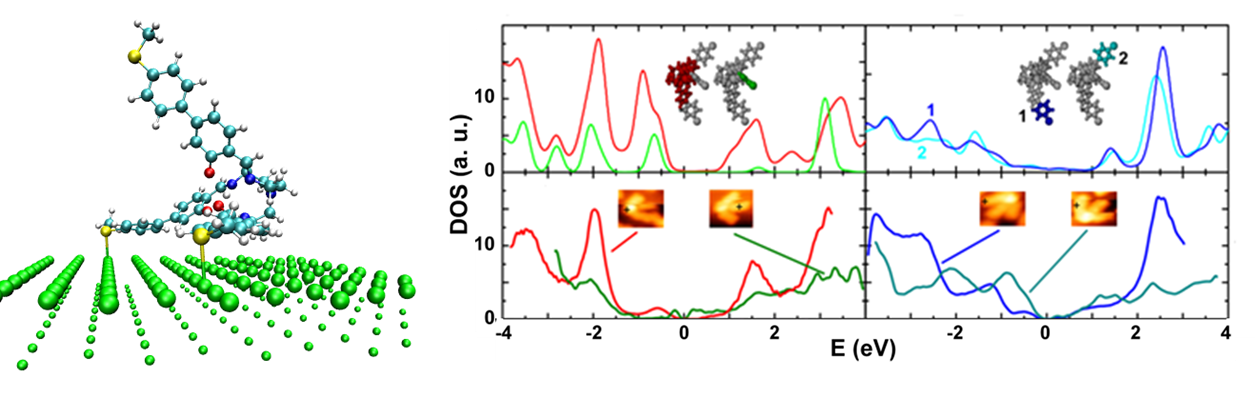
M. Lukas, K. Doessel, A. Schramm, O. Fuhr, C. Stroh, M. Mayor, K. Fink, and H. v. Loehneysen
A Tripodal Molecule on a Gold Surface:Orientation-Dependent Coupling and Electronic Properties of the Molecular Legs
ACS Nano 7, 6170 (2013)
Magnetic and catalytic properties of polynuclear transition metal complexes
Karin Fink
In the collaborative research project SFB/TRR 88 3MET researchers at the Technische Universität Kaiserslautern and at the Karlsruher Institute of Technology collaborating in the fields magnatism, catalysis and spectroscopy. The research is focussed on bi and trinuclear transition metal complexes and clusters.
We investigate the electronic structure of transition metal compounds with wave function based ab initio methods. We are particularly interested in compounds where first order spin orbit coupling is present.
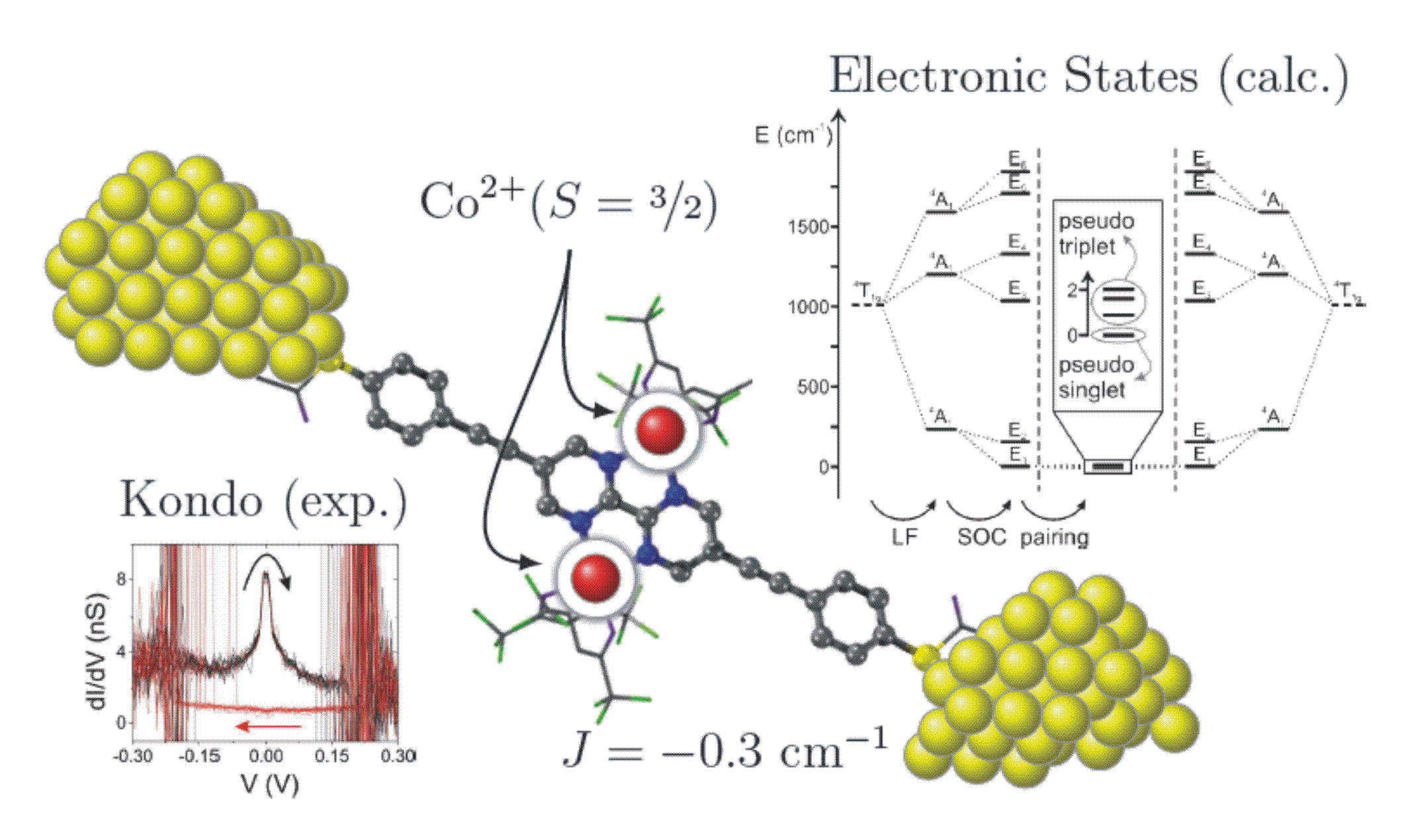
Switching of a coupled spin pair in a single-molecule junction
S. Wagner, F. Kisslinger, S. Ballmann, F. Schramm, R. Chandrasekar, T. Bodenstein, O. Fuhr, D. Secker, K. Fink, M. Ruben, H. B. Weber
Nature Nanotechnology 2013, 8, 575
For the calculation of the magnetic data we use a modified CASSCF (complete active space self consistent field) approach and include the spin orbit coupling of the low lying electronic states. Magnetic exchange coupling constants, zero field splittings, and the g-factors can be extracted from the calculated data.