
The Multiscale Materials Modeling and Virtual Design group at the Institute for Nanotechnology helps to interpret and design experiments in the areas of nanoscale structure formation and function. We develop and apply methods for multi-scale simulations of nanoscale materials and devices, with particular emphasis on nanoscale electronics (single molecule electronics, organic electronics) and carbon based systems with emphasis on graphene and carbon nanotubes. A key goal of our method is the development of predictive scale-bridging methods for materials design and discover. As multi-scale methods cover a several different space and/or time scales it is important to interlink different formalisms to construct an accurate model, technically implemented in workflows. In the MMM@HPC project (www.multiscale-modelling.eu) we have developed a transferable and extendable platform for multiscale materials simulations based in UNICORE workflow engine.

Physical vapor deposition is used widely to produce organic electronics devices, in particular for OLEDs and in recent years have seen a number of surprising observations, such as deposition-induced emitter orientation and built-in electrostatic potentials in thin organic films. In this paper, we compare two different modeling approaches, which both aim to simulate the physical vapor deposition (PVD) process for small organic molecules; A molecular dynamics approach, based on a classical bead-spring force field and time integration and a Metropolis Monte Carlo approach, where the intramolecular degrees are limited to the torsional rotations. To analyze the resulting structures, we calculate the density and radial distribution functions (RDF) of all films. We observe a good agreement for the RDFs, but an approximately 10% higher density for the films generated by the molecular dynamics approach. Additionally, we investigate the anisotropic nature of such morphologies by calculating the ordinary and extra-ordinary refractive index for each material. Finally, we calculate electron and hole mobilities with an Kinetic Monte Carlo protocol.
Organic Electronics, DOI: 10.1016/j.orgel.2022.106439
This roadmap presents the transformational research ideas proposed by “BATTERY 2030+,” the European large-scale research initiative for future battery chemistries. A “chemistry-neutral” roadmap to advance battery research, particularly at low technology readiness levels, is outlined, with a time horizon of more than ten years. This roadmap should be seen as an enabling complement to the global battery roadmaps which focus on expected ultrahigh battery performance. Lithium-ion batteries will soon approach their performance limits, but through this “chemistry neutral” approach a generic toolbox transforming the way batteries are developed, designed and manufactured, will be created.
Adv. En. Mat., DOI: 10.1002/aenm.202102785Modeling and simulation of materials have become indispensable to complement experiments in materials design, aiding researchers in selecting the most promising materials for experimental studies or providing insights inaccessible by experiment. This often requires multiple simulation tools, so methods and tools are needed to enable extensive-scale simulations with streamlined execution of all tasks within a complex simulation protocol. These methods should allow rapid prototyping of new protocols and proper documentation of the process. We present an overview of the benefits and challenges of workflow engineering in virtual material design and a selection of prominent scientific workflow frameworks used for the research in the BATTERY 2030+ project.
Adv. En. Mat., DOI: 10.1002/aenm.202102638
A completely new and modular receptor design strategy based on microporous hybrid materials is presented yielding zeolite-based artificial receptors (ZARs) which reversibly bind the neurotransmitters serotonin and dopamine with unprecedented affinity and selectivity even in saline biofluids. ZARs are thermally and chemically robust, can be readily prepared at gram scales and their applicability for the label-free monitoring of important enzymatic reactions, for (two-photon) fluorescence imaging, and for high-throughput diagnostics in biofluids is demonstrated.
Adv. Mat., DOI: 10.1002/adma.202104614
Graphene is inherently sensitive to vicinal dielectrics and local charge distributions, a property that can be probed by the position of the Dirac point in graphene field-effect transistors.Mmetal–organic frameworks can be tailored to selective adsorption of specific molecular species. Here, a selective ethanol sensor is demonstrated by growing a surface-mounted metal–organic framework (SURMOF) directly onto graphene field-effect transistors (GFETs). Tailoring multiple SURMOFs to adsorb specific gases on an array of such devices thus generates a versatile, selective, and highly sensitive platform for sensing applications.
Adv. Mat., DOI: 10.1002/adma.202103316
Organic semiconductors are widely used in prominent applications such as organic light‐emitting diode displays and organic solar cells. The state of the art of predictive simulation methods is discussed, including machine learning, to complement experimental research in the identification of novel materials in the vast available chemical space, and their potential is illustrated with a few prominent recent applications.
Adv. Mat., DOI: 10.1002/adma.201808256
Proton‐conducting molecules in the pores of metal–organic frameworks change their conductivity upon photoswitching the host framework. In this work, irradiation with light (green) causes trans–cis isomerization of the azobenzene components of the framework, switching the molecular interaction and the conductivity of the triazole guest molecules (center). The sample is mounted on interdigitated gold electrodes, which are used for measuring the conductivity.
Adv. Mat., DOI: 10.1002/adma.201706551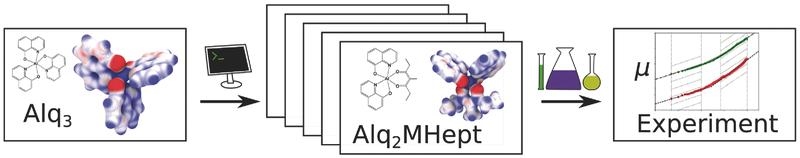
The viability of a multiscale simulation approach to rationally design organic semiconductors with improved electron mobility is demonstrated. Novel materials with tailored electronic properties are designed for which an improvement of the electron mobility by three orders of magnitude is predicted and experimentally confirmed.
Adv. Mat, DOI: 10.1002/adma.201703505