
Polymers
Polymers belong to a versatile class of functional molecules and have an essential role in nature and for diverse technical applications. Due to the enormous variety of monomers (building blocks) available for polymer structuring, they possess a widespread application possibilities and a vast tunability of the desired properties. Here, both the chemical composition of a macromolecule and its spatial arrangement, as well as the connectivity and dimensionality, are of key importance. We employ scale-bridging computational methods to analyze and increasingly predict the structure and functional properties of diverse macromolecules, i.e. starting from the quantum mechanical calculations of individual polymer building blocks, but ranging all the way to mesoscopic model of polymer networks and their functional properties. Moreover, we work on the continuous improvement of modelling techniques of soft matter in close collaboration within SFB 1176 Collaborative Research Centre. This permits tailoring of the soft matter functionality to the specific applications.
Challenges and limits of mechanical stability in 3D direct laser writing
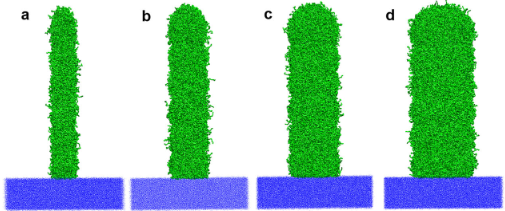
Direct laser writing, using ultrashort laser pulses, is an effective technique for fabrication of complex 3D polymer networks, but it is difficult to obtain a time-resolved microscopic picture of the printing process in operando. To close this gap, a molecular dynamics simulation approach is presented to model direct laser writing and investigate the effect of writing condition and aspect ratio on the mechanical properties of the printed polymer network. We show that writing conditions provide a possibility to tune the mechanical properties and an optimum writing condition can be applied to fabricate structures with improved mechanical properties.
Tacticity dependence of single chain polymer folding
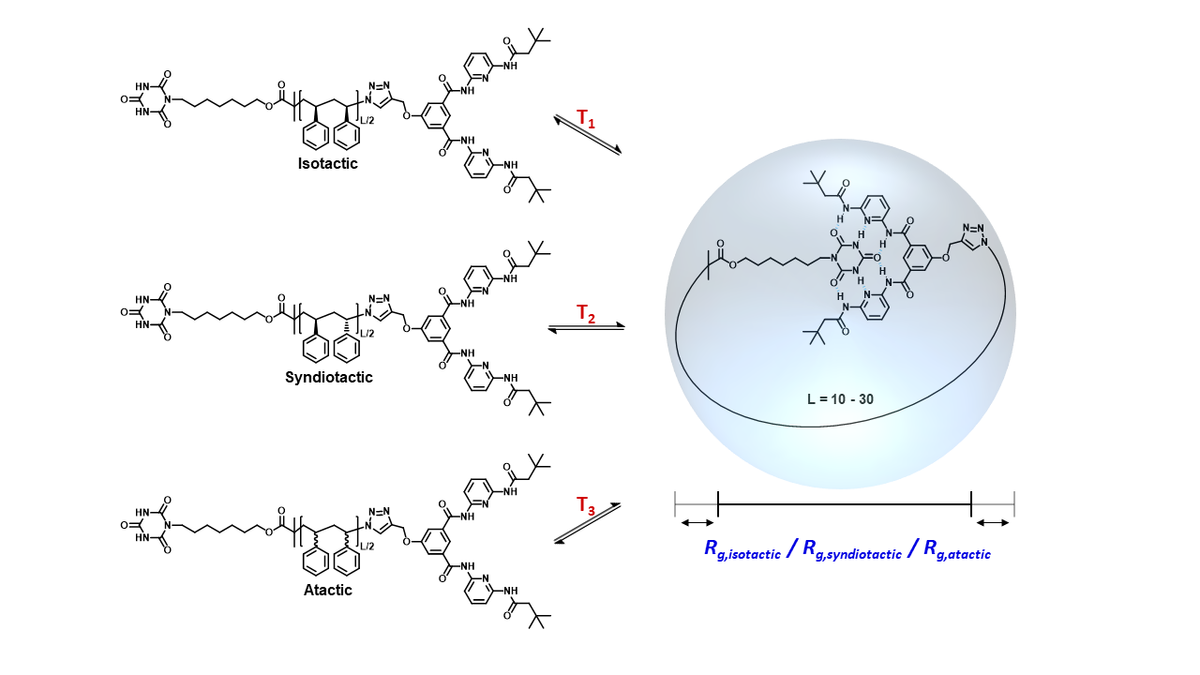
Modelling of reversible single chain polymer self-assembly: from the polymer towards the protein limit
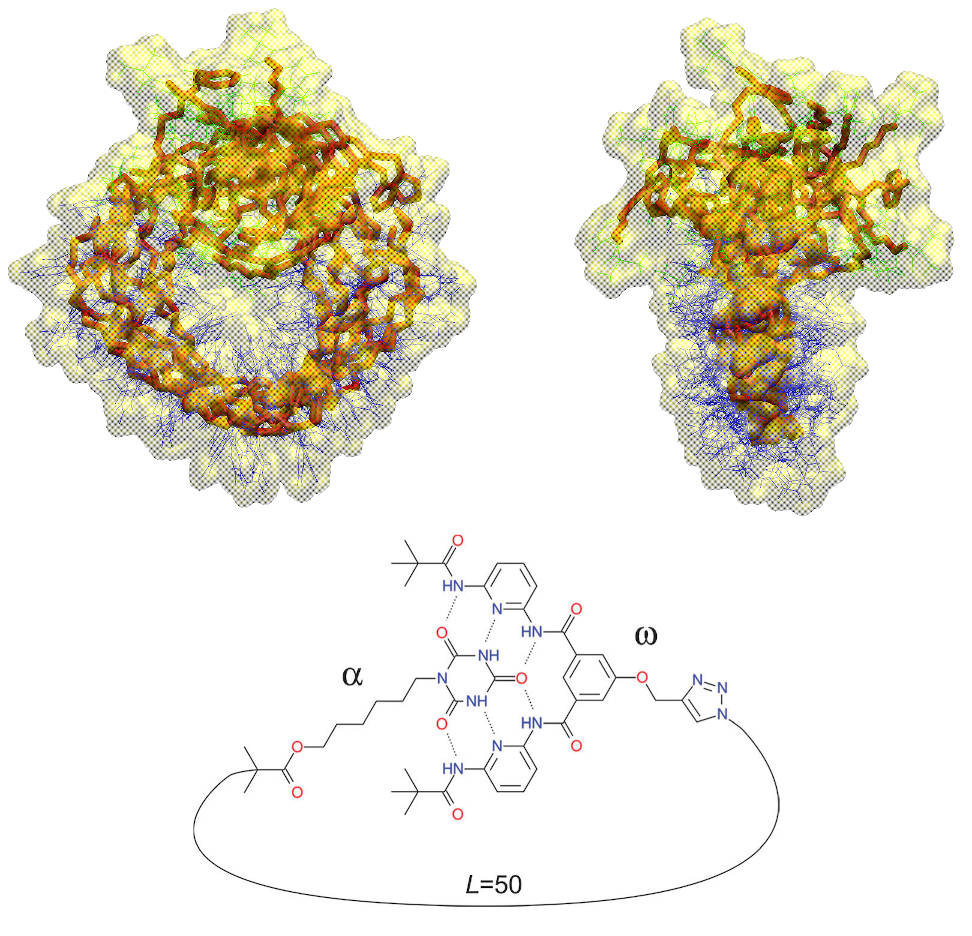
What are the conditions on the polymer chain enabling its ‘‘folding’’ not just into a collapsed conformation, but into a single ‘‘unique’’ conformation? How does the folding process differ from that of proteins, where folding is driven by a multitude of interactions optimized by evolution, while polymer folding is driven by a few engineered interactions? Which level of accuracy is required in terms of polydispersity and placement of the point folding units to achieve a dominant unique conformation in the ‘‘folded’’ ensemble? We use all-atom simulations to fully characterize the thermodynamic equilibrium of reversible single-chain point folding and to predict at what chain length ‘‘folding’’ into a defined geometry of the single chain units is expected to occur.
Selective Dispersion of Large-Diameter Semiconducting Single-Walled Carbon Nanotubes with Pyridine-Containing Copolymers
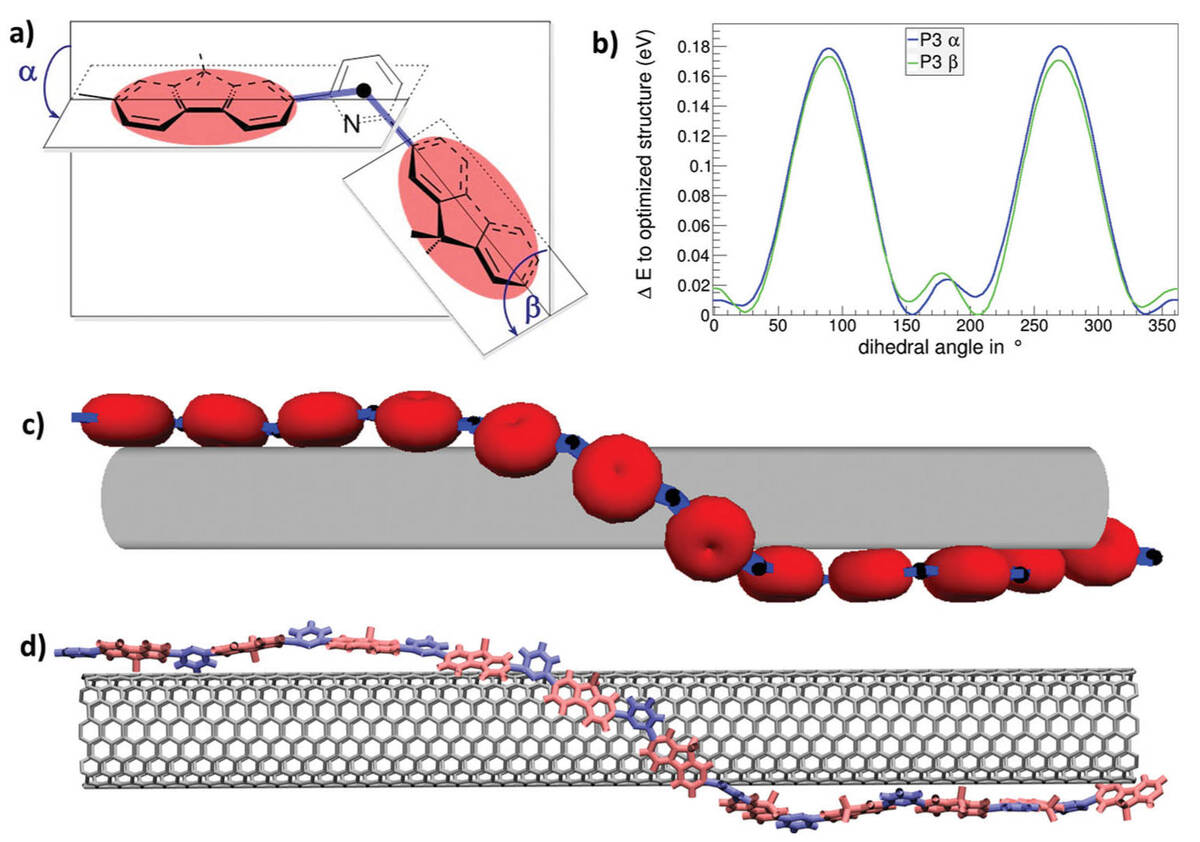
To obtain high performance electronic, optoelectronic and photonic devices, based on single-walled carbon nanotubes (SWNTs), the purification of SWNTs is required. We simulated the processes taking place at the interface between SWNTs and pyridine-containing copolymer during SWNTs purification by a newly designed hybrid coarse-grain model combining density functional theory and geometrical calculation.
The developed model provides an impressive match with the experimentally found SWNTs diameter selectivity to the specific copolymer and yields insights into the polymer wrapping processes with an unprecedented level of detail for large diameter SWNTs.
Effect of Geometrical Structure, Drying, and Synthetic Method on Aminated Chitosan-Coated Magnetic Nanoparticles Utility for HSA Effective Immobilization
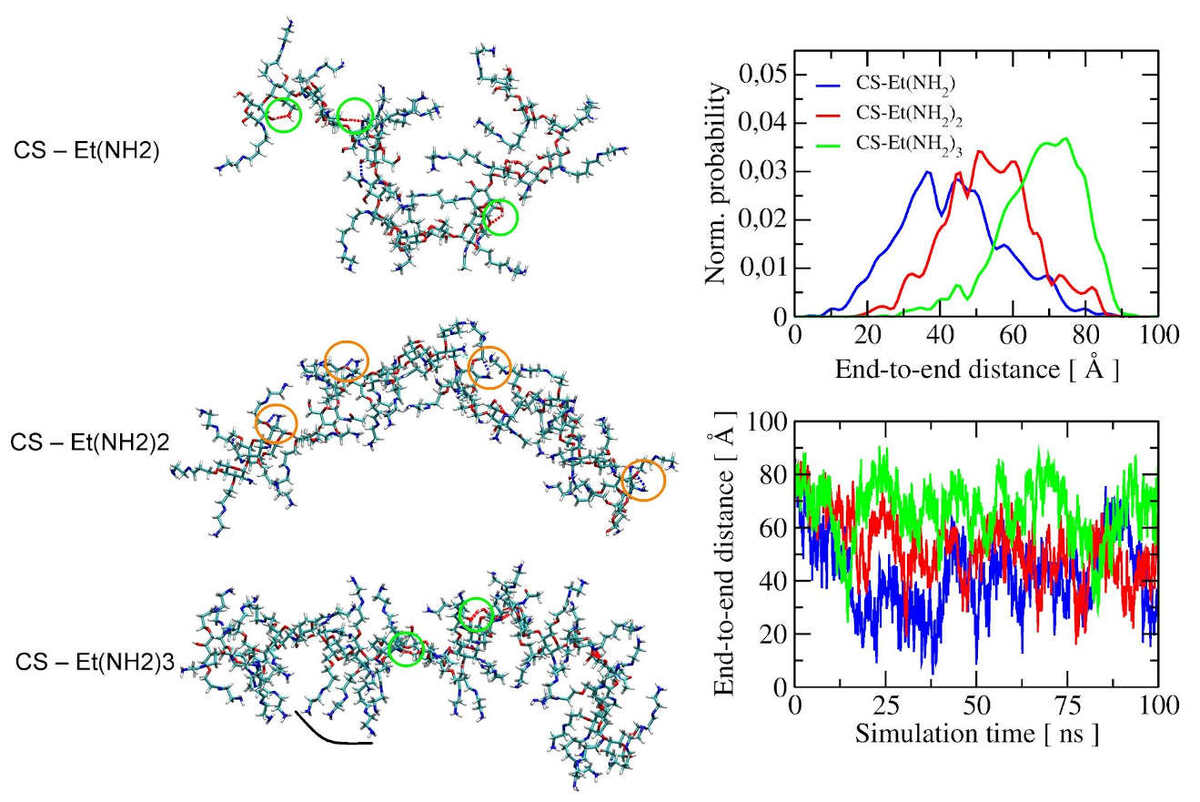
Binding of a drug with human serum albumin (HSA) is one of the basic pharmacokinetic parameters determined for a drug, indicating its pharmacological activity. Different nanocarriers are proposed for efficient immobilization of HSA for better drug determination. Here, we report three types of aminated chitosan-coated magnetic nanoparticles with a different content of amino groups, long distanced from the surface, as a promising binding platform for effective HSA immobilization and protein separation from the supernatant, simply by applying a magnet. We apply molecular dynamics simulations to investigate the influence of the geometry and conformational variability of the shell structure of aminated chitosan on the ability to bind HSA and to explain significant binding differences of the Fe3O4-CS-Et(NH2)2 polymer.